La nouvelle réglementation sur les dispositifs médicaux: l’essentiel en bref
Une nouvelle réglementation européenne relative aux dispositifs médicaux est entrée en vigueur le 26 mai 2021. Le règlement 2017/745, également appelée MDR pour « Medical Device Regulation », remplace les anciennes directives 90/385/CEE et 93/42/CEE. Ce règlement impose la mise en conformité et le respect de certaines obligations à tous les acteurs économiques de la filière et s’applique à tous les dispositifs médicaux, y compris ceux déjà commercialisés.
Objectifs de la MDR
- Garantir le maintien de la sécurité, de la fiabilité et de la qualité des dispositifs médicaux
- Renforcer la transparence de l’information pour les utilisateurs et les professionnels de santé
- Renforcer la traçabilité, la surveillance après commercialisation et la vigilance
La MDR créé un cadre réglementaire solide, transparent et durable qui est reconnu au niveau international. Elle est directement applicable par tous les pays de l’Union Européenne et ne doit pas être transposée en droit national.
Exigences de la MDR
Cette nouvelle réglementation redéfinit le statut et les responsabilités des parties prenantes. Alors que la législation précédente ne s’appliquait qu’aux fabricants, représentants autorisés, assembleurs, importateurs et distributeurs, la MDR accorde le statut d’opérateur économique et des responsabilités à tous les acteurs du secteur :
- Fabricants, assembleurs, reconditionneurs, re-processeurs
- Mandataires
- Importateurs
- Distributeurs
- Acteurs de la vente à distance et in-house
- Etablissements de santé
La MDR amènent notamment les changements suivants (liste non-hexaustive)
- Introduction d’exigences essentielles et de procédures renforcées pour la conception et la fabrication des dispositifs médicaux.
- Exigences accrues pour l’obtention du marquage CE ainsi que pour les organismes notifiés en charge de l’évaluation de la conformité et de la certification. Les organismes notifiés ont un cahier des charges élargi et sont placés sous contrôle européen.
- Désignation d’une/de personne(s) chargées de veiller au respect de la réglementation (PCVRR). Celle-ci a pour mission de s’assurer de la conformité des dispositifs médicaux, fournir la documentation technique et mettre à jour la déclaration de conformité UE.
- Vigilance renforcée sur le report et le traitement des incidents, la surveillance après mise sur le marché et la gestion des données issues du suivi clinique après commercialisation.
- Mise en place d’une base de données européenne consacrée aux dispositifs médicaux (EUDAMED). La commission européenne prévoit son lancement au deuxième trimestre 2027.
EUDAMED permettra aux utilisateurs et professionnels de santé d’accéder à des informations sur les dispositifs médicaux commercialisés en Europe, de connaître les incidents déclarés ainsi que l’avancée des investigations cliniques. Un identifiant unique (IUD) sera attribué à chaque dispositif médical pour améliorer sa traçabilité.
- Mise en place d’un système d’identification unique des dispositifs médicaux (UDI : Identifiant Unique du produit) qui figurera sur l’emballage du produit sous la forme d’un code barre ou d’un code QR. Il permettra une traçabilité très précise du dispositif, notamment utile en cas de d’incident ou de rappel de produits. Un calendrier séparé existe et prévoit une mise en application au deuxième
trimestre 2029.
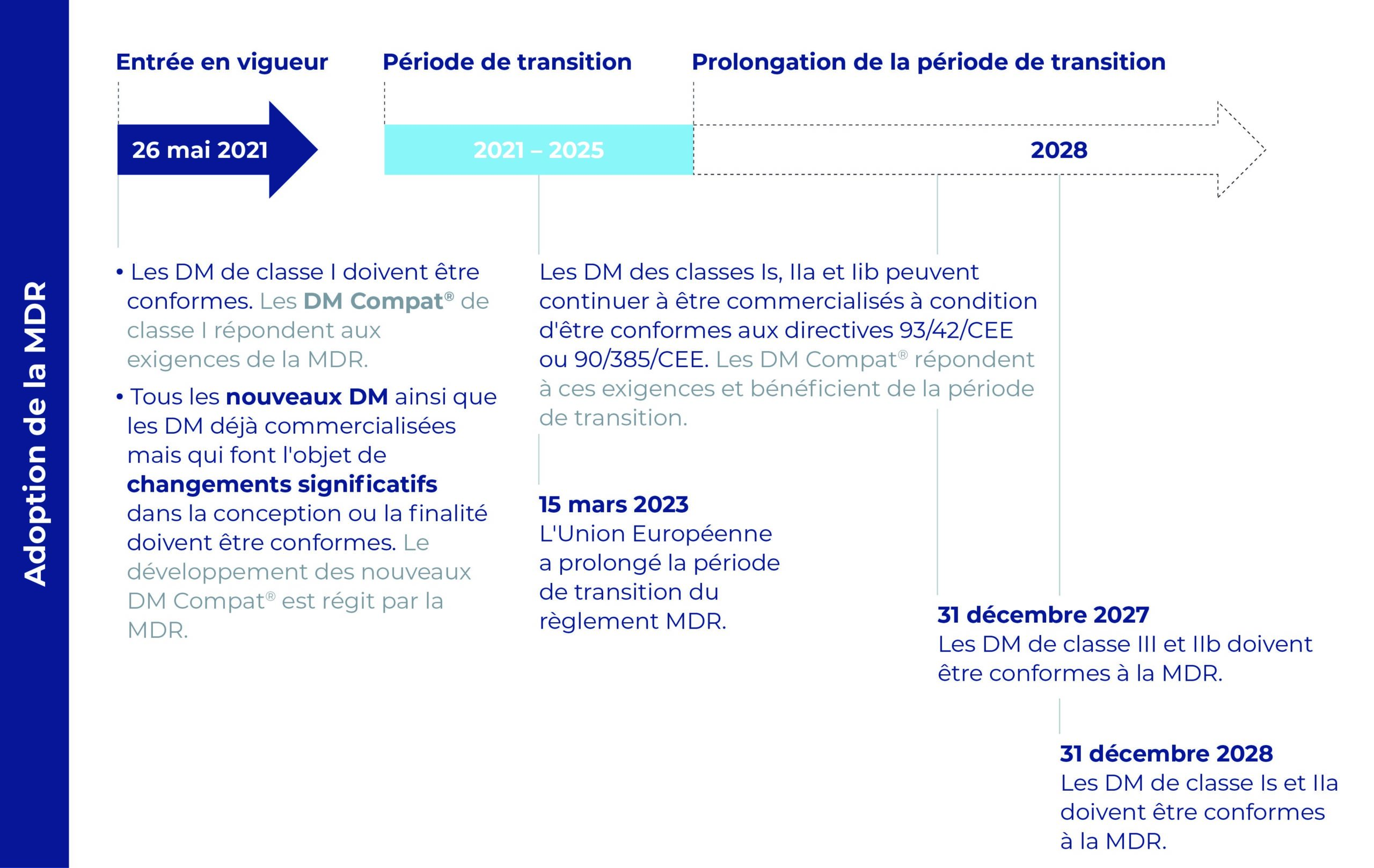
Le calendrier de la nouvelle réglementation
La gamme Compat est prête !
Tous les produits Compat sont mis en conformité avec les exigences de la MDR aux dates applicables :
- 26 mai 2021 pour les dispositifs de classe I
- A la date d’expiration du certificat CE, ou au plus tard :
– le 31 décembre 2027 pour les dispositifs de classe IIb
– le 31 décembre 2028 pour les dispositifs de classes Is et IIa.
Pour plus d’informations, contactez-nous.

