Medical Device Regulation (MDR) at a glance
A new European regulation on medical devices came into force on 26 May 2021. Regulation 2017/745, also known as the Medical Device Regulation or simply “MDR”, replaces the previous directives 90/385/EEC and 93/42/EEC. It requires that all industry stakeholders comply with certain obligations, and applies to all medical devices, including those already on the market.
Aims of the MDR
- Ensure the continued safety, reliability and quality of medical devices
- Reinforce transparency of information for healthcare users and professionals
- Reinforce traceability, post-marketing surveillance and vigilance
The MDR creates a sound, transparent, durable regulatory framework that is recognised worldwide. It is directly applicable in all European Union (EU) countries and must not be transposed into national law.
Requirements of the MDR
The new regulation redefines the status and responsibilities of stakeholders. Whereas previous legislation only applied to manufacturers, authorised representatives, assemblers, importers and distributors, the MDR grants “economic operator” status and responsibilities to all of the sector’s stakeholders:
- Manufacturers, assemblers, re-packagers, re-processors
- Agents,
- Importers
- Distributors
- Remote and in-house sales representatives
- Healthcare institutions
The MDR brings about several changes, including but not limited to:
- It introduces essential requirements and strengthens procedures for the design and manufacture of medical devices.
- It increases the requirements for obtaining CE marking as well as for the notified bodies that assess conformity and certification. These notified bodies now have expanded specifications and are placed under EU control.
- It provides that a designated person be appointed to ensure compliance with the regulation. This person is responsible for the conformity of the medical devices, supplies technical documentation and updates the EU declaration of conformity.
- It strengthens vigilance regarding incident reporting and handling, post-marketing surveillance and management of data from post-marketing clinical follow-up.
- It creates a European database specifically for medical devices (EUDAMED). The European Commission expects this database to be launched in Q2 2027.
EUDAMED will enable healthcare users and professionals to access information on medical devices marketed in Europe, review reported incidents and track the progress of clinical investigations. A unique device identifier (UDI) will be assigned to each medical device to improve its traceability.
- Set-up of a unique identification system for medical devices (UDI: Unique Device Identifier), which will appear on the product package in the form of a barcode or QR code. It will enable accurate tracking of medical devices, which is useful if an incident occurs or a product is recalled. There is a separate calendar with plans for implementation in Q2 2029.
The calendar for the new regulation and repercussions for Compat Medical Devices (MD)
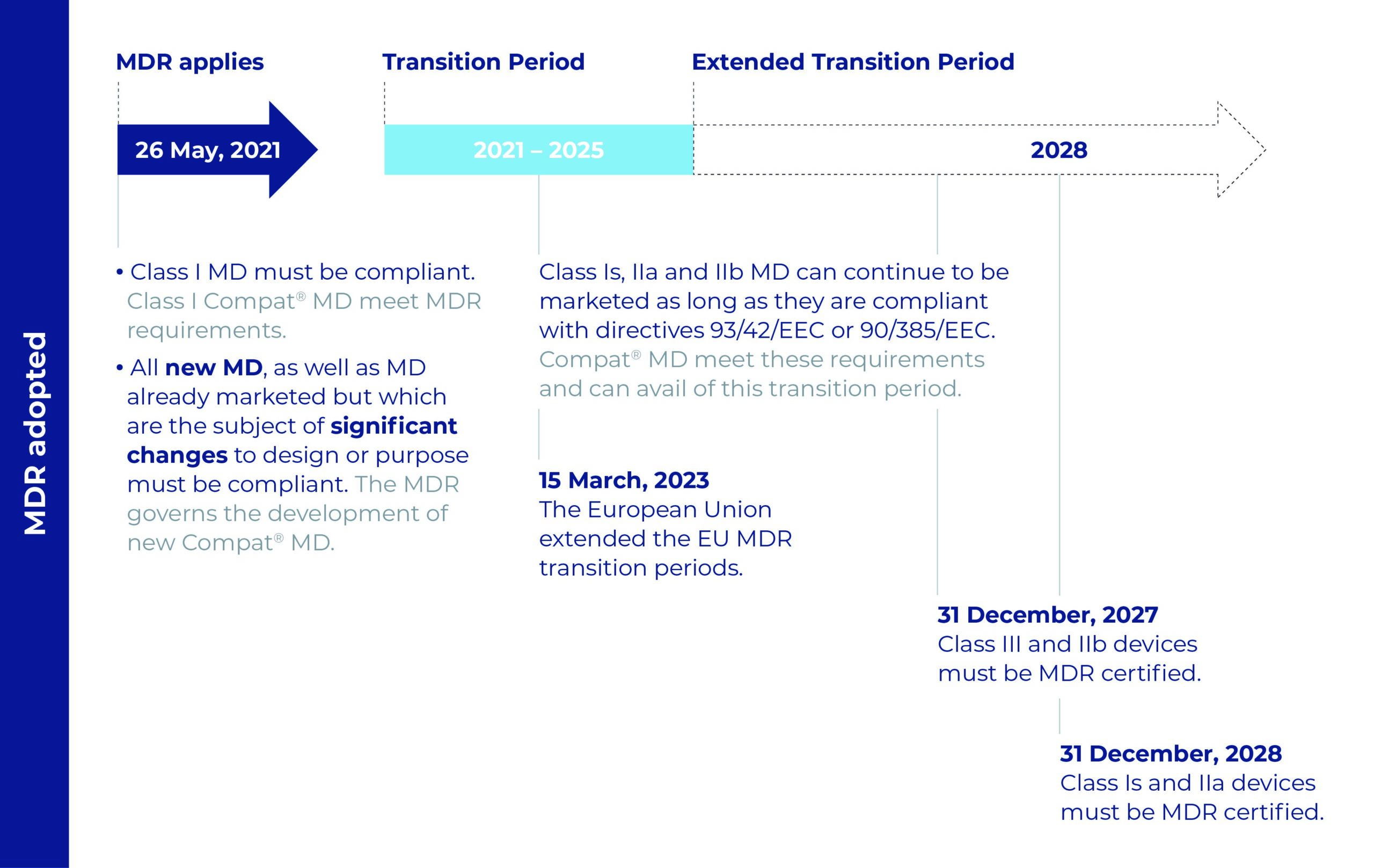
The Compat range is ready!
All Compat products are being made compliant with MDR requirements by the applicable dates:
- 26 May 2021, for class I medical devices
- By the date the MDD CE certifi cate expires or no later than:
– 31 December 2027 for class IIb
– 31 December 2028 for class Is and IIa.
For more information, please contact us

